The human genome encodes the blueprint of life, but the function of the vast majority of its nearly three billion bases is unknown. The Encyclopedia of DNA Elements (ENCODE) project has systematically mapped regions of transcription, transcription factor association, chromatin structure and histone modification. These data enabled us to assign biochemical functions for 80% of the genome, in particular outside of the well-studied protein-coding regions. Many discovered candidate regulatory elements are physically associated with one another and with expressed genes, providing new insights into the mechanisms of gene regulation. The newly identified elements also show a statistical correspondence to sequence variants linked to human disease, and can thereby guide interpretation of this variation. Overall, the project provides new insights into the organization and regulation of our genes and genome, and is an expansive resource of functional annotations for biomedical research.

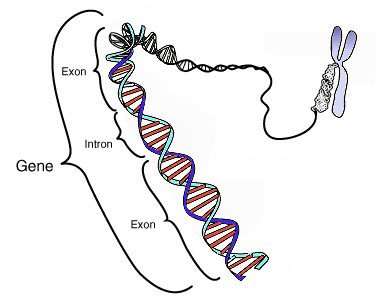
The ENCODE project provides information on the human genome far beyond that contained within the DNA sequence — it describes the functional genomic elements that orchestrate the development and function of a human. The project contains data about the degree of DNA methylation and chemical modifications to histones that can influence the rate of transcription of DNA into RNA molecules (histones are the proteins around which DNA is wound to form chromatin). ENCODE also examines long-range chromatin interactions, such as looping, that alter the relative proximities of different chromosomal regions in three dimensions and also affect transcription. Furthermore, the project describes the binding activity of transcription-factor proteins and the architecture (location and sequence) of gene-regulatory DNA elements, which include the promoter region upstream of the point at which transcription of an RNA molecule begins, and more distant (long-range) regulatory elements. Another section of the project was devoted to testing the accessibility of the genome to the DNA-cleavage protein DNase I. These accessible regions, called DNase I hypersensitive sites, are thought to indicate specific sequences at which the binding of transcription factors and transcription-machinery proteins has caused nucleosome displacement. In addition, ENCODE catalogues the sequences and quantities of RNA transcripts, from both non-coding and protein-coding regions.
The papers are "open access" there are a large number of them so if you are interested download them. Maybe start with
Ecker JR et al. Genomics: ENCODE explained