Stoffels et al. Fibronectin aggregation in multiple sclerosis lesions impairs remyelination. Brain. 2013;136:116-31. doi: 10.1093/brain/aws313.
Background: Remyelination following central nervous system demyelination is essential to prevent axon degeneration. However, remyelination ultimately fails in demyelinating diseases such as multiple sclerosis. This failure of remyelination is likely mediated by many factors, including changes in the extracellular signalling environment.
Methods and results: Here, we examined the expression of the extracellular matrix molecule fibronectin on demyelinating injury and how this affects remyelination by oligodendrocytes progenitors. In toxin-induced lesions undergoing efficient remyelination, fibronectin expression was transiently increased within demyelinated areas and declined as remyelination proceeded. Fibronectin levels increased both by leakage from the blood circulation and by production from central nervous system resident cells. In chronically demyelinated multiple sclerosis lesions, fibronectin expression persisted in the form of aggregates, which may render fibronectin resistant to degradation. Aggregation of fibronectin was similarly observed at the relapse phase of chronic experimental autoimmune encephalitis, but not on toxin-induced demyelination, suggesting that fibronectin aggregation is mediated by inflammation-induced demyelination. Indeed, the inflammatory mediator lipopolysaccharide induced fibronectin aggregation by astrocytes. Most intriguingly, injection of astrocyte-derived fibronectin aggregates in toxin-induced demyelinated lesions inhibited oligodendrocyte differentiation and remyelination, and fibronectin aggregates are barely expressed in remyelinated multiple sclerosis lesions.
Conclusions: Therefore, these findings suggest that fibronectin aggregates within multiple sclerosis lesions contribute to remyelination failure. Hence, the inhibitory signals induced by fibronectin aggregates or factors that affect fibronectin aggregation could be potential therapeutic targets for promoting remyelination.
Methods and results: Here, we examined the expression of the extracellular matrix molecule fibronectin on demyelinating injury and how this affects remyelination by oligodendrocytes progenitors. In toxin-induced lesions undergoing efficient remyelination, fibronectin expression was transiently increased within demyelinated areas and declined as remyelination proceeded. Fibronectin levels increased both by leakage from the blood circulation and by production from central nervous system resident cells. In chronically demyelinated multiple sclerosis lesions, fibronectin expression persisted in the form of aggregates, which may render fibronectin resistant to degradation. Aggregation of fibronectin was similarly observed at the relapse phase of chronic experimental autoimmune encephalitis, but not on toxin-induced demyelination, suggesting that fibronectin aggregation is mediated by inflammation-induced demyelination. Indeed, the inflammatory mediator lipopolysaccharide induced fibronectin aggregation by astrocytes. Most intriguingly, injection of astrocyte-derived fibronectin aggregates in toxin-induced demyelinated lesions inhibited oligodendrocyte differentiation and remyelination, and fibronectin aggregates are barely expressed in remyelinated multiple sclerosis lesions.
Conclusions: Therefore, these findings suggest that fibronectin aggregates within multiple sclerosis lesions contribute to remyelination failure. Hence, the inhibitory signals induced by fibronectin aggregates or factors that affect fibronectin aggregation could be potential therapeutic targets for promoting remyelination.
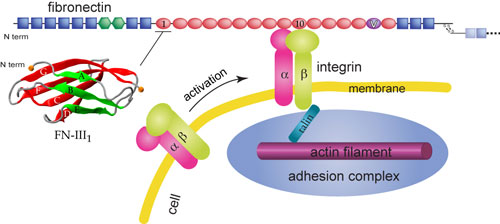
Recently we talked about Fibrogen affecting lesion formation, this report looks at fibronectin which is a blood product that is deposited in MS and EAE lesions. This can influence astrocyte function and in this study they implicate the production of fibronectin aggregates as a factor that influences remyelination.